A Fully Validated, Multi-Kilogram “cGMP” Synthesis of “MDMA”. Jay B Nair et al
A Fully Validated, Multi-Kilogram “c-G-M-P” Synthesis of “M-D-M-A”.
Jay B Nair, Linda Hakes, Berra Yazar-Klosinski, and Kathryn Paisner.
ACS Omega. 2022, number 7, pages 900 to 907.
ABSTRACT: “M-D-M-A” is increasingly used in clinical research, but no “c-G-M-P”, Current Good Manufacturing Practice, process has yet been reported. We describe here the first fully validated “c-G-M-P” synthesis of up to 5 kilograms, about thirty thousand patient doses, of “M-D-M-A” in a four-step process beginning with a non-controlled starting material. The overall yield was acceptable, 41 to 53 percent, over four steps, and the chemical purity of the final product was excellent, exceeding 99.9 percent of the peak area by “H-P-L-C” in each of the four validation trials. The availability of “c-G-M-P” compliant “M-D-M-A” will facilitate ongoing clinical trials and provide for future therapeutic use, if encouraging results lead to “F-D-A” approval.
INTRODUCTION.
Interest in the clinical utility of psychedelic compounds has increased dramatically in recent years. Although medical usage of these substances, in tandem with psychotherapy, was briefly and controversially explored, in the 1950s and 1960s, increased regulatory oversight and social disapprobation effectively eliminated such research until the late 1990s, when tentative efforts to revive it commenced. Promising early results very slowly stimulated additional engagement, both experimentally and culturally, provoking recent regulatory shifts that have further stimulated engagement by making research chemicals more accessible and expanding the permissible scope of clinical studies.
This second wave of so-called psychedelic studies more expansively includes compounds like entactogen 3, 4-methylenedioxymethamphetamine (“M-D-M-A”). Like traditional psychedelics, “M-D-M-A” had previously enjoyed a brief period of encouraging early-stage exploration, in the 1970s and 1980s, which was similarly curtailed by social and regulatory backlash.
In contrast to psychedelics like LSD and psilocybin, however, the addition of “M-D-M-A” to the U.S. Drug Enforcement Administration’s (DEA) Schedule One appeared to be largely related to “M-D-M-A”’s popularity as an illicit “party drug,” rather than to significant concerns regarding either contemporary research efforts or its therapeutic utility. Indeed, in clinical trials conducted since the U S Food and Drug Administration (“F-D-A”) and DEA first granted research approval, in 2004, “M-D-M-A” has shown promise as a psychotherapeutic aid for patients suffering from “P-T-S-D”, autism-related social anxiety, and alcoholism.
As the research environment grows steadily more supportive of clinical exploration, and as successful clinical trials open the door for fully approved treatments, the need for pharmaceutically acceptable “M-D-M-A” continues to expand. To ensure that patients receive safe, effective drugs, the manufacture of pharmaceutical substances is closely regulated by the “F-D-A”, under a structure called Current Good Manufacturing Practice (“c-G-M-P”). These rules delineate standards for every aspect of the manufacturing process, including facility design, establishment and documentation of operating procedures, process monitoring, and chemical analysis. Because only small samples of each pharmaceutical batch are submitted for destructive quality control testing, a well-controlled manufacturing process is the best-known way to ensure that all drugs distributed to consumers are of predictably high quality, consistency, and efficacy. “c-G-M-P” compliant synthetic processes are typically developed for drug candidates in tandem with progressing clinical trials.
Unlike many drug candidates, “M-D-M-A” enjoyed a robust synthetic history prior to receiving any serious consideration as a pharmaceutical substance. “M-D-M-A” was first synthesized by Merck, in 1912, as an intermediate to the styptic compound methyl-hydrastitine. Scientists periodically explored its pharmacological effects over the intervening half-century, both at Merck and in the United States Army, but “M-D-M-A” does not appear in either the patent or the chemical literature again until 1960, when Biniecki and Krajewski published a synthesis identical to Merck’s in Poloniae Pharmaceutica, it is unlikely that they were aware of the Merck patent.
This synthetic route proceeded via hydrobromination of the natural product safrole (Scheme 1, TWO), yielding the Markovnikov adduct (Scheme 1, THREE), which was then converted to “M-D-M-A” using methylamine in methanol. A variety of synthetic approaches from methyl piperonyl ketone (Scheme 1, FOUR), which was commercially available at the time, and which can be easily prepared either from safrole typically via Wacker oxidation, or piperonal (Scheme 1, FIVE) typically by reducing its nitroalkene derivative with iron in hydrochloric acid, were summarized by Shulgin, in 1986. A novel approach from piperonal, via Curtius rearrangement, was reported by Schulze, in 2010, and a handful of asymmetric syntheses of (S)-”M-D-M-A”, some relying on alternate starting materials, have also appeared in the literature (Scheme 1).
Clandestine chemists preparing “M-D-M-A” for the black market have additionally developed a number of synthetic routes from readily available starting materials like catechol (Scheme 1, 7), eugenol (Scheme 1, 8), isosafrole (Scheme 1, 9), and piperine (Scheme 1, 10), though most still approach “M-D-M-A” through a safrole or (less frequently) piperonal intermediate. These synthetic methods often rely on chemicals readily available to ordinary consumers, in an effort to circumvent controlled substance precursor regulations.
Most of these clandestine syntheses are well documented, both by anonymous chemists, in online forums, and by forensic scientists, who often identify clandestine production methods by their distinct impurity profiles.
To date, none of the synthetic explorations into “M-D-M-A” appear to have considered “c-G-M-P”s. While intended for pharmaceutical production, Merck’s early investigations occurred less than a decade after the “F-D-A” was founded, and well before its “c-G-M-P” rules were developed. Some clandestine labs reliably produce large quantities of high-quality “M-D-M-A”; however, these facilities necessarily operate outside of regulatory frameworks and certainly do not report or document “c-G-M-P”-compliant procedures. Most other synthetic explorations of “M-D-M-A” have been geared toward the production of “M-D-M-A” as a research chemical, usually for small-scale studies in animals or for forensic analysis (Figure one).
Indeed, prior to a recently completed Phase 3 trial for “P-T-S-D”, it is likely that few even contemplated a need for “c-G-M-P”-compliant “M-D-M-A”. As a Schedule One substance, it officially had no recognized medical utility up until now. As a well-known compound with a lengthy history in the public domain and a short treatment regimen, it also had little apparent commercial value.
RESULTS.
We report here the first “c-G-M-P” synthesis of “M-D-M-A” and its hydrochloride salt (“M-D-M-A”, ”Hydrochloride”), which is used in pharmaceutical formulations. In this fully validated, four-stage process, up to 5 kilogram of “M-D-M-A”, ”Hydrochloride” was reproducibly synthesized, with an overall yield of 41.8 to 54.6 percent and a minimum purity of 99.4 percent (weight by weight) by “H-P-L-C” assay. Over a minimum of four consecutive trials, for each stage, the established targets for yields and impurity profiles were achieved, and, in most cases, exceeded.
Chemical impurities in the final product (“M-D-M-A”, ”Hydrochloride”) averaged 0.04 percent of the total peak area, by “H-P-L-C”, and no single impurity ever exceeded 0.03 percent of the total peak area. Of all of the organic solvents used in the production process, only isopropanol (Class 3, 409 to 509 parts per million), tetrahydrofuran (Class 2, less than 7 parts per million), methanol (Class 2, less than 6 parts per million), and n-heptane (Class 3, less than 67 parts per million) were detected in the final product, all in concentrations well below the permitted daily exposure (PDE) per “F-D-A” guidance. The scale and reliability of this “c-G-M-P” process will improve access to “M-D-M-A” for ongoing and future clinical trials, and potentially for licensed therapeutic use, pending “F-D-A” approval.
DISCUSSION.
Increased demand for pharmaceutical-grade “M-D-M-A” encouraged us to develop a “c-G-M-P”-compliant production process, both to supply our own Phase three clinical trials, for “P-T-S-D”, and to ameliorate existing supply constraints for the broader research community. While large-scale clandestine production is common, to the best of our knowledge, no multi-kilogram synthesis of pharmaceutical-grade “M-D-M-A” has yet been reported in the literature. We therefore needed to develop a practicable synthetic route while simultaneously addressing “c-G-M-P” requirements.
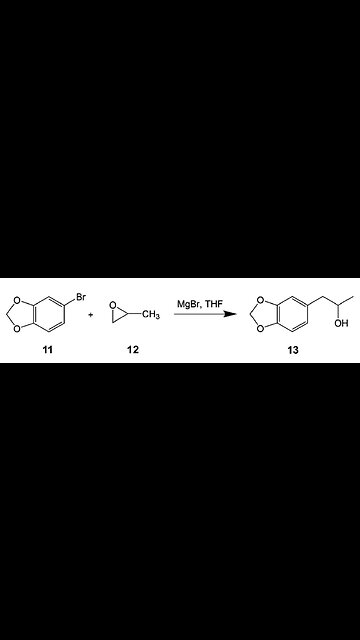
“M-D-M-A” is not a particularly complex molecule, and many synthetic pathways have been reported. Most begin from either safrole or piperonal, which are highly regulated and consequently difficult to obtain; for the sake of convenience and efficiency, we elected to avoid these. We identified 5-bromo-1, 3-benzodioxole (Compound 11), which does not appear on any geopolitical entity’s list of controlled substance precursors, as a useful starting material for our synthesis. The 1, 3-benzodioxole moiety appears in a variety of natural products, including oils, spices, and traditional plant-based medicines. Many compounds containing this structural feature are known to interact with cytochrome P450 enzymes in mammals, producing a range of clinically notable effects, both pharmacologically useful and neurotoxic. Compound 11 is synthesized via the bromination of benzodioxole with NBS; analysis of multiple batches, from a range of suppliers, indicated that the only significant impurities present in the batch are 5,6-dibromo-1,3-benzodioxole and succinimide, which is insoluble in Compound 11 and consequently present in only very trace amounts. We additionally screen for the presence of 4-bromo-1, 3-benzodioxole, which would likely present separation challenges during production, but we have never observed this isomer in the starting material. At the levels observed, neither of the two significant impurities interfered with the downstream chemistry.
Compound 11 has been previously used in at least two (reported) approaches to “M-D-M-A”: as a starting material in the asymmetric synthesis of (S) ”M-D-M-A”, through a protected aziridine intermediate, and as a precursor to safrole, via Grignard reaction with allyl bromide (Scheme 2). Instead of approaching “M-D-M-A” conventionally, via safrole, we elected to generate a 2-propanol substituent via a ring opening addition between the same aryl Grignard reagent used to synthesize safrole, above, and 1, 2-propylene oxide (Compound 12), which is both inexpensive and readily available.
Reactions of Grignard reagents and epoxides are well-known, but, to the best of our knowledge, this particular synthetic pathway has not previously been used to produce “M-D-M-A”. Our familiarity with this type of reaction made us optimistic that scale-up would proceed smoothly, and it did. Although Grignard formation is slow, the bulk reaction can be expedited via initiation with a small amount of previously prepared Grignard reagent. The 5, 6-dibromo-1, 3-benzodioxole impurity present in the starting material does not undergo Grignard formation and is removed during workup as part of the organic layer. The workup at this stage was quite efficient, and distillation via a wiped-film evaporator, two to three passes, yielded 1-(3,4-methylenedioxy-phenyl)-2-propanol (Chemical 13) in excess of 96 percent chemical purity by “H-P-L-C”. The adjusted yield, based on “H-P-L-C” assay, was 79.22 to 87.39 percent (weight by weight) over five trials.
The next three steps relied on well-known synthetic transformations. Compound 13 was oxidized to methyl piperonyl ketone (Scheme1, 4) with a biphasic (DCM, H2O) TEMPO, Potassium Bromide, bleach reagent system, which was followed by aqueous workup and filtration to remove remaining solids. The solvent was removed via a rotatory evaporator, and the crude product was of sufficient purity to proceed to the next process stage, without an additional purification step (100.2 to 108.2 percent yield over four trials; 84.9 to 90.01 percent weight by weight by “H-P-L-C” assay). Stage 3, reductive amination of Compound 4, was accomplished with aqueous methylamine and Sodium Hydroxide, Sodium Borohydride. Workup was somewhat complex, using an acid and base treatment to remove the vast majority of impurities, followed by acidification with “Hydrochloride” in isopropanol which yielded 71.6 to 75.8 percent “M-D-M-A”, ”Hydrochloride” (Compound 14), over eight trials, with chemical purity exceeding 99.26 percent of peak area, by “H-P-L-C”.
Recrystallization in isopropanol (Stage 4) yielded 85 to 86.2 percent of a white, crystalline solid, with a minimum purity of 99.95 percent by “H-P-L-C” and a minimum assay of 99.40 percent (weight by weight), also by “H-P-L-C” (Table 1).
“M-D-M-A”, ”Hydrochloride” was previously known to form one major crystal form (Form One) and at least four hydrates that incorporate 0.25 to 1 waters of hydration. Our polymorphic screening process identified two new anhydrous crystal forms (Forms Two and Three) and established Form One as the most stable of the three. Form Two can be reproducibly produced from a variety of alcoholic solvents, as well as in the presence of ethyl acetate and an ethereal antisolvent. Unlike Form Three, which spontaneously converted to Form One after 2.5 weeks at ambient conditions, and could not be reproduced, Form Two is shelf stable, though it will convert to Form One under competitive equilibration conditions. Interestingly, both Form One and Form Two reversibly convert into the known monohydrate; upon dehydration, the monohydrate formed from Form One will revert back to Form One, and the monohydrate formed from Form Two will revert back to Form Two. If crystallized from a concentrated aqueous solution with no form memory, the monohydrate will thermally dehydrate exclusively into Form One. X-ray powder diffraction spectra for all three forms are shown in Figure 2.
To maintain compliance with “c-G-M-P” regulations, all reagents were visually inspected and tested, prior to use. Conformance to established specifications was documented, reagents were labeled with identifying raw material numbers, and these identifying numbers were recorded whenever a reagent was used, in-process. Organic reagents were typically confirmed by FT-IR, as well as by other methods specific to their chemical identity and various process needs, for example Karl Fischer titration to establish water content, and so on, in accordance with established procedures. Inorganic reagents were confirmed by appropriate chemical identification tests. Reagents that failed to meet all established specifications were not used at any stage of the process.
Another concern for “c-G-M-P” manufacturing is the presence of residual solvents, which must be below solvent-specific concentration thresholds defined in USP 467. The limits set for residual solvent concentrations are based on anticipated daily exposure to a pharmaceutical product. In clinical use,
“M-D-M-A” is never recommended for daily, or even regular, consumption; instead, it is ingested during a small number of therapy sessions, spread over weeks or months. Nevertheless, our monograph utilizes the USP 467 PDE limits as acceptance criteria, and our process yielded residual solvent concentrations significantly below these limits, over four consecutive validation trials (Table 2). The limit of detection for all tested solvents was 1 part per million; solvents detected in concentrations below the quantitation limit were reported as such.
In addition to meeting residual solvent concentration limits, “c-G-M-P” pharmaceuticals must have acceptable impurity profiles.
Any single impurity exceeding 0.1 percent must be both characterized and quantified.
Over four trials, our process yielded “M-D-M-A”, ”Hydrochloride” with chemical purity in excess of 99.9 percent of peak area by “H-P-L-C”; no single impurity ever exceeded 0.05 percent of the total peak area. While impurity characterization was consequently not required, we routinely screened for two known impurities (Figure 3), both of which were generated via low level electrophilic addition during the Stage 2 oxidation of Chemical 13.
Chlorination was only significant when the bleach was overcharged, and the reaction conditions used in Stage 2 prevent this. Bromination, which also increased with excess bleach, was a more significant side reaction, but it was successfully minimized using Potassium Bromide in catalytic, rather than stoichiometric, quantities. Neither impurity was ever found in excess of 0.03 percent of the total peak area, by “H-P-L-C”, in any of the four Stage 4 validation trials.
Heavy metal impurities in finished pharmaceutical products are also an area of potential concern. As with residual solvents, “c-G-M-P”-compliant limits are established with the assumption that a medication will be consumed on a daily basis, a usage pattern that we do not anticipate will ever be in effect for clinically administered “M-D-M-A”. Nevertheless, we used the oral daily dose PDEs from USP 232 when determining acceptability parameters. As shown in Table 3, the greatest quantifiable amount of any heavy metal impurity was 97 percent less than the permissible daily intake limit, and most were well below that level.
To validate this “c-G-M-P” process, each stage was successfully completed at the 8 kilogram scale (based on the starting charge of benzodioxole) at least four consecutive times, in accordance with the documented procedures. All reagents, products, intermediates, common impurities, and, as required, reaction end points were validated using “c-G-M-P”-compliant analytical methods, some of which were specifically developed for this synthetic process. Any deviations from the documented procedures or parameters were noted, and the anticipated impact on the final product, if any was characterized. No documented deviation appeared to affect either the final product or the outcome of the Stage 4 recrystallization step, which yielded remarkably consistent results throughout the validation process (Table 1). We are confident that our “c-G-M-P” protocols are sufficient to reliably produce enough pharmaceutically acceptable “M-D-M-A” to meet expanding research and therapeutic needs.
EXPERIMENTAL SECTION.
General. Reactions were performed using commercially available raw materials and solvents. Unless otherwise stated, all commercially obtained reagents were qualified prior to use and then used as received. Reactions were conducted in a 50 Liter reaction vessel. The small-scale production of the Grignard reagent, used to initiate the bulk reaction, was conducted in a 2 Liter reactor fitted with a reflux condenser. A Huber Unistat was used for temperature control and logging. In-process analysis was conducted by “H-P-L-C”, with supplemental H one NMR analysis used to quantify residual solvent content during evaporation steps. A wiped-film evaporator was used for distillation. All processes were conducted under nitrogen (target: less than 5 percent O2).
Residual solvent testing was performed on an Agilent J and W DB-624 HRGC column (60 meters times 0.32 millimeters, 1.80 micron film thickness).
Stage 1, the Synthesis of 1-(3, 4-Methylenedioxyphenyl)-2-propanol. Grignard Formation.
Into a 2 Liter vessel was charged 16.6 grams of magnesium turnings (0.68 mole, 1.1 equivalents), followed by 500 milliliters of THF (181 parts per million H2O by KF titration) at 20 degrees C. Stirring was initiated after the introduction of THF, and the vessel was heated to a gentle reflux. To the vessel was then charged 125 grams of 5-bromo-1, 3-benzodioxole, 0.62 moles, 1 equivalent, chemical purity greater than 98.70 percent by “H-P-L-C”) in two unequal portions. The first portion weighed 6.3 grams and was stirred for 12 hours at a gentle reflux until an exotherm was observed. Following this initiation step, the remaining 118.4 grams was added, dropwise, to the reaction vessel, and the resultant Grignard solution was stirred at reflux for 40 minutes and then cooled to 25 degrees C.
Into a separate 50 Liter reaction vessel was charged 1.06 kilograms of magnesium turnings (44 moles, 1.1 equivalent) and 32 Liters of THF.
The suspension was stirred and then heated to a gentle reflux.
To the reaction vessel was then added 400 grams of 5-bromo-1, 3-benxodioxole (2.0 moles), followed by 400 milliliters of the small batch Grignard solution described above.
Reflux was maintained. After 5 minutes, a significant increase in the reflux rate was observed in the glass condenser, indicating initiation.
While maintaining reflux, 7.6 kilograms of 5-bromo-1, 3-benxodioxole (38 moles) was then added to the reaction vessel, using a dropping funnel, and the batch was stirred at a gentle reflux for 40 minutes.
Addition of Propylene Oxide.
The bulk Grignard solution was cooled to 10 degrees Celsius, and 128.8 grams of copper iodide (1.5 moles, 0.4 equivalent) was added to the 50 Liter vessel. A solution of 2.5 Liter (plus, minus) propylene oxide (37 moles, 0.93 equivalents) in 2.5 Liters of THF was then added to the reaction vessel while maintaining the temperature at 0 to 10 Celsius. The container and dropping funnel were rinsed with an additional 800 milliliters of THF, which was then added to the 50 Liter reaction vessel. The batch was stirred for 40 minutes at 5 to 20 degrees C, forming a dark brown solution and a crystalline suspension.
Completion analysis performed by “H-P-L-C” confirmed the reaction end point (0.30 percent 5-bromo-1,3- benzodioxole; target limit was less than or equal to 1 percent).
The batch was then divided into two 20.4 Liter portions for workup. For each portion, 5.45 Liters of a 10 percent (weight per weight) sodium chloride solution, followed by 1.37 Liters of acetic acid, was added to the 50 Liter reaction vessel while maintaining the temperature at 10 to 25 degrees. The half-batch portion was then transferred from carboy into the reaction vessel while ensuring that the temperature remained below 40 degrees. Following this addition, the half-batch portion was stirred at 30 to 40 degrees for 45 minutes; then, the pH was adjusted to less than 5 by sequential addition of three 200 milliliters of aliquots of acetic acid. The batch was allowed to settle, and the aqueous layer was removed. 8.2 Liters of n-heptane were then charged into the 50 Liter reaction vessel, followed by an additional 8 Liters of the sodium chloride solution. The batch was stirred and allowed to settle, and the aqueous layer was again removed. The dark brown organic layer was filtered over a vacuum, using a plate filter with a 11 micron filter mesh. Following workup, the two half-batches were combined, and the solvent was removed in a 20 Liter rotatory evaporator. The crude yield was 7442.7 grams and analysis by H one NMR revealed 2.5 percent residual THF, n-heptane not detected; target limit is less than or equal to 10 percent total amount of both solvents.
The crude product was charged with 1488.5 grams of PEG400, 0.2 equivalent weight per weight and mixed to ensure homogeneity. This mixture was then distilled at 150 to 185 degrees and 0.1 to 1.5 milli bars, using a wiped-film evaporator. Two passes yielded 6293.2 grams of a pale yellow oil (94.2 percent yield; 96.44 percent area, 89.78 percent weight per weight by “H-P-L-C”).
Stage 2.
Oxidation to 1-(3, 4-Methylenedioxyphenyl)-propan-2-one. A 50 Liter reaction vessel was charged with 2760.1 grams of crude 1-(3, 4-methylenedioxyphenyl)-2-propanol from Stage 1 (88.56 percent weight per weight by “H-P-L-C” assay; active charge is 2444.3 grams, 13.6 moles, 1 equivalent) and 9780 milliliters of dichloromethane at 10 to 25 degrees. Stirring was initiated, and 178.5 grams of potassium bromide was added, followed by 233.2 grams of TEMPO (0.11 equivalents). The batch was cooled to 0 degrees, and 7280 milliliters (60 percent) of a solution of sodium hydrogen carbonate (0.25 equivalent) in 12120 milliliter of bleach, 1.6 equivalent, diluted to 12.5 percent weight per volume) was added, dropwise, while stirring efficiently and maintaining the temperature at minus 10 to 10 degrees. A 1 milliliter sample was then removed, for analysis by “H-P-L-C”, and four additional 610 milliliter (5 percent) of aliquots of the Sodium Bicarbonate bleach solution were then added, dropwise, to the reaction vessel. A sample was collected after each addition, and “H-P-L-C” analysis was used to monitor the reaction progress. After the fourth aliquot was added, 1.61 percent of Stage 1 starting material remained (target limit is less than 5 percent). Stirring was halted, and the layers were allowed to settle. The layers were separated, and the organic layer was returned to the 50 Liter vessel.
For workup, the organic layer was cooled to 0 Celsius, and 4890 milliliters of a 12 percent (weight per weight) solution of aqueous sodium hydrosulfite was added while maintaining the temperature at 0 to 10 degrees. The reaction mixture was then warmed to 19.5 degrees and stirred for 15 minutes. The layers were separated, and the organic layer was returned to the 50 Liter reaction vessel. Then, 4900 milliliters of freshly prepared 0.5 Molar aqueous Sodium Hydroxide was added, and the reaction mixture was stirred for 15 minutes. The layers were separated, and the brown organic layer was returned to the 50 Liter reaction vessel. To this was added 4900 milliliters of 11 percent (weight per weight) aqueous Sodium Chloride, followed by 98 milliliters of concentrated Hydrochloric acid 36 percent weight per weight aqueous solution. After stirring for 15 minutes at 18.5 degrees, the layers were separated, and the organic layer was returned to the reaction vessel. Two more washes, the first with another 4900 milliliters of the 11 percent Sodium Chloride solution, the second with 4900 milliliters of a saturated Sodium Chloride solution were completed, following the same procedure. The organic layer was filtered over a Buchner funnel fitted with a filter cloth rinsing with 500 milliliters of DCM and then transferred to a 20 Liter rotatory evaporator. The solvent was removed under vacuum, yielding 2442.1 grams of a yellow-to brown oil (101.0 percent crude yield; 94.52 percent peak area, 89.80 weight per weight by “H-P-L-C”).
Stage 4. Recrystallization of “M-D-M-A”·”Hydrochloride”.
To a 50 Liter reaction vessel was added 4107.3 grams of crude “M-D-M-A”, ”Hydrochloride” and 41 Liters of 2-propanol. The batch temperature was increased to 67.2 degrees, while stirring, and the mixture was then stirred for 30 minutes at 67.2 degrees until all of the solids dissolved. Stress tests had demonstrated stability for 72 hours at 70 to 80 degrees proving the thermal stability of “M-D-M-A”, ”Hydrochloride”.
The batch was then transferred through a 1.2 micron in-line filter capsule, using positive pressure, to a clean, 50 Liter reaction vessel, fitted with a jacket that had been preheated to 66.1 Celsius.
In this new reaction vessel, the batch was cooled to 55.3 degrees, over the course of 90 minutes. Then, 41.1 grams of “M-D-M-A”, ”Hydrochloride” Form 1 seed crystal (0.18 moles, 0.008 equivalents) was added, and the batch was stirred at the same temperature for 30 minutes. The batch was cooled to 15.2 degrees at a rate of 3 degrees per hour and then stirred at this temperature for an additional 10 hours.
The white suspension was removed from the mother liquor via vacuum filtration over a filter plate fitted with a filter cloth and then washed with 8220 milliliters of 2-propanol. The filter cake was transferred to a drying oven and dried under vacuum (140 millibars) for 19 hours at 56.6 degrees. The collected “M-D-M-A”, ”Hydrochloride” was a white solid weighing 3548.3 grams (85.5 percent yield; 99.95 percent peak area, 99.64 percent weight by weight by “H-P-L-C”). No single impurity exceeded 0.02 percent of the peak area by “H-P-L-C”, and residual solvents, methanol, less than six parts per million; 2-propanol, 490 parts per million, were found to be within the target range.
-
 6:49
6:49
Russell Brand
23 hours ago"HE'S A RUSSIAN PLANT!" CNN Loses It ON AIR!
119K162 -
 13:10
13:10
The Rubin Report
1 day agoWhy the Real Challenge Is Just Beginning | Jordan Peterson
62.9K23 -
 1:02:55
1:02:55
Tactical Advisor
5 hours agoBuilding a Truck Gun -Battle Hawk Build of the Month | Vault Room Live Stream 017
42K -
 42:41
42:41
Athlete & Artist Show
4 hours ago $1.41 earnedSeason 5 Episode 3 LIVE
26.1K2 -

I_Came_With_Fire_Podcast
12 hours agoThe US GOVERNMENT is PLANNING a UAP FALSE FLAG ATTACK
22.3K6 -
 18:10
18:10
Sideserf Cake Studio
6 hours ago $0.92 earnedIs This the ULTIMATE Cake Smashing Moment?
24.9K2 -
 12:51
12:51
Misha Petrov
19 hours agoTrump KICKS OUT Zelenskyy After HEATED White House Meeting!
23.9K60 -
 16:39
16:39
Tactical Considerations
1 day ago $0.90 earnedWatchtower Apache Double Stack 1911 Made Me Question Everything?
19.2K -
 16:20
16:20
T-SPLY
7 hours agoCNN Meltdown Over Zelesnky Disrespecting Donald Trump And JD Vance
16.2K20 -
 8:39
8:39
Silver Dragons
1 day agoGold & Silver Price KEEP DROPPING 👀
11.6K7